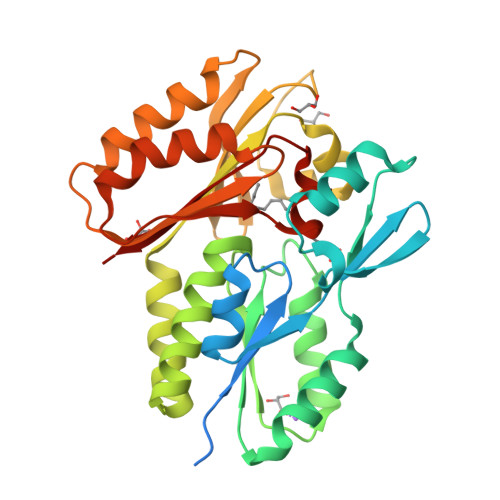
ペプチド
抗体
色素試薬
PROTAC
バーチャルスクリーニング
TargetMolキット
Cell Counting Kit-8 (CCK-8)
Inhibitor Cocktails
天然化合物
フェノール類
アルカロイド類
フラボノイド類
阻害剤
血管新生
アポトーシス
オートファジー
細胞周期・チェックポイント
クロマチン・エピジェネティック
細胞骨格シグナル
DNA損傷・DNA修復
内分泌・ホルモン
GPCR/Gタンパク質
免疫・炎症
JAK・STATシグナル
MAPK

Cytokines and Growth Factors
• Cytokines
• Growth Factors
Receptor Proteins
• Fc Receptors
• Cytokines and Growth Factor Receptors
CD Proteins
• T Cell CD Proteins
• B Cell CD Proteins
Immune Checkpoint Proteins
• Co-inhibitory Immune Checkpoint Proteins
• Co-stimulatory Immune Checkpoint Proteins

 参考文献
参考文献 Calculator
Calculator